Introduction
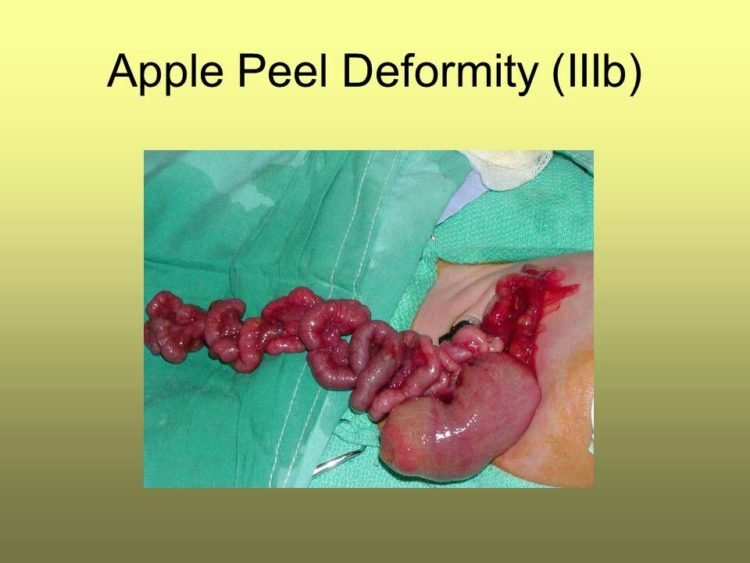
Apple-peel intestinal atresia, also known as type IIIb or Christmas tree intestinal atresia, is a rare form of small bowel atresia in which the duodenum or proximal jejunum ends in a blind pouch and the distal small bowel wraps around its vascular supply in a spiral resembling an apple peel. Often a vast segment of small bowel is absent.
While apple-peel intestinal atresia is usually reported as an isolated malformation, it has been associated with malrotation. Additionally, syndromic association has been reported with microcephaly and ocular anomalies.
Jejunal atresia is a birth defect in a newborn characterized by partial or complete absence of the membrane connecting the small intestines to the abdominal wall (the mesentery). It causes a portion of the small intestines (the jejunum) to twist around an artery that supplies blood to the colon (the marginal artery).
This leads to an intestinal blockage or “atresia.” Common symptoms in the newborn include feeding difficulties, failure to thrive, vomiting bile (a yellowish-green fluid), abdominal swelling, and/or absence of bowel movements after birth. It typically occurs sporadically in people with no family history of the condition; however, more than one family member can rarely be affected, suggesting that there may be a genetic component in some cases. Jejunal atresia is typically treated with surgery.
We report a case of mixed type IIIb and type IV intestinal atresia. The baby underwent exploratory laparotomy where he was found to have the entire distal bowel supplied by one blood vessel that appeared to have undergone an antenatal volvulus. The volvulus was untwisted and upon further exploration the patient was noted to have twelve distal atresias. We performed a total of six primary small bowel anastomoses. The baby did not require any further procedures and did well post-operatively. We conclude that patients with this congenital abnormality can be treated with primary anastomoses, and when appropriate, without the need for enterostomy or stent.
Intestinal atresia is a malformation where there is a narrowing or absence of a portion of the intestine. This defect can either occur in the small or large intestine.
Malrotation of gut is a serious congenital abnormality of GI tract as it predisposes to duodenal obstruction and midgut volvulus which can lead to ischaemic necrosis of small bowel. Underdiagnosis of midgut volvulus carries a high mortality rate. During fetal life the gut begins as a straight midline tube which elongates and rotates around SMA of 270 degrees in anticlockwise direction to form the entire adult bowel. Malrotation is a generic term used to describe any variation in the position of intestines and is in itself not symptomatic. Most patients (70%) have isolated malrotation of the bowel but there is an increased incidence of associated duodenal stenosis and atresia [1]. We present a case of malrotation with atresia of jejunum and ileum.
Atresia of small intestine is a special form of intestinal atresia with absence of mesentery, which is most likely due to an intrauterine intestinal vascular accident. Newborns are usually preterm infants with low birth-weights, that encounter feeding difficulties (including vomiting with initial feeds, which may later worsened and the abdomen becomes progressively distended) as well as failure to thrive. Affected children present disrupted bowel loops assuming a spiral configuration resembling an ‘apple peel’ and may have less than half of the normal length of the small bowel and a physiologically short bowel. Atresia of small intestine is characterized by jejunal atresia near the ligament of Treitz, foreshortened bowel, and a large mesenteric gap. The bowel distal to the atresia is precariously supplied. Atresia of small intestine may be a manifestation of cystic fibrosis (see this term). The most important cause of mortality is short bowel syndrome (see this term), encountered in 65% of cases.
ejunoileal atresias are complete obstructions of the jejunum or ileum Caused by arterial and venous abnormalities in the mesentery of the bowel of the fetus More common in the jejunum, equal in males and females and may be single or multiple in nature Associated anomalies are less common than in duodenal atresias
Intestinal atresia and stenosis involve narrowing or closure of the intestine. Food is blocked from passing through the intestine, which prevents normal feeding and intestinal function. Intestinal atresia and stenosis usually involve the small intestine, but can affect any part of the gastrointestinal tract. Intestinal stenosis is a partial obstruction that causes the center opening of the intestine to become narrower, while intestinal atresia is a complete closure of the intestine.
Intestinal atresias are a common cause of bowel obstruction in newborns. Jejunoileal obstruction was found in 46% of the intestinal atresias. The classification of jejunoileal atresias is defined by 4 types of lesions. Jejunal atresia (JA) is generally considered to result from intrauterine vascular disruptions in a segment of the developed intestine. Destructive events such as volvulus, herniation, constriction, and intussusception have been observed during surgery in patients with JA. Early and prompt diagnosis and appropriate treatment have improved these patients’ outcomes. Rapid diagnosis is important in the treatment of this anomaly.
Symptoms
Signs and symptoms of jejunal atresia vary but may include:[3][4]
- Feeding difficulties
- Failure to thrive
- Vomiting bile (a bitter-tasting, yellowish-green fluid)
- Abdominal swelling, especially the upper middle part just below the breast bone
- Absence of bowel movements after birth
This table lists symptoms that people with this disease may have. For most diseases, symptoms will vary from person to person. People with the same disease may not have all the symptoms listed. This information comes from a database called the Human Phenotype Ontology (HPO) . The HPO collects information on symptoms that have been described in medical resources. The HPO is updated regularly. Use the HPO ID to access more in-depth information about a symptom.
The different types of intestinal atresia are named after their location:
- Duodenal atresia – malformation of the duodenum, part of the intestine that empties from the stomach
- Jejunal atresia – malformation of the jejunum, the second part of the intestine extending from the duodenum to the ileum
- Ileal atresia – malformation of the ileum, the lower part of the small intestine
- Colon atresia – malformation of the colon
Duodenal atresia has a strong association with Down syndrome.[1] It is the most common type, followed by ileal atresia.
An inherited form – familial multiple intestinal atresia – has also been described. This disorder was first reported in 1971.[2] It is due to a mutation in the gene TTC7A on short arm of chromosome 2 (2p16). It is inherited as an autosomal recessive gene and is usually fatal in infancy.
Cause
Jejunal atresia occurs when the membrane that attaches the small intestines to the abdominal wall (called the mesentery) is partially or completely absent. As a result, a portion of the small intestines (the jejunum) twists around an artery that supplies blood to the colon (the marginal artery). This leads to an intestinal blockage or “atresia.”
Scientists suspect that it may be a consequence of disruption of blood flow in the developing fetus, leading to the death of cells and tissue in the affected area (necrosis). There may be various reasons that blood flow becomes disrupted. Because jejunal atresia rarely occurs in more than one family member, there may be a genetic component or predisposition in some cases.
The most common cause of non-duodenal intestinal atresia is a vascular accident in utero that leads to decreased intestinal perfusion and ischemia of the respective segment of bowel. This leads to narrowing, or in the most severe cases, complete obliteration of the intestinal lumen.
In the case that the superior mesenteric artery, or another major intestinal artery, is occluded, large segments of bowel can be entirely underdeveloped. Classically, the affected area of bowel assumes a spiral configuration and is described to have an “apple peel” like appearance; this is accompanied by lack of a dorsal mesentery. Ileal atresia can also result as a complication of meconium ileus.
Diagnosis
In some cases, jejunal atresia may be diagnosed before birth on a prenatal ultrasound or fetal MRI.[1][5] This is helpful because infants can be treated promptly after birth, reducing the risk of complications. Ultrasound findings that may suggest intestinal atresia include dilated loops of bowel, hyperechoic bowel (it appears brighter than it should), and accumulation of fluid (ascites). Because prenatal ultrasound is not always accurate, X-rays and imaging studies with contrast should be obtained after birth to confirm the diagnosis.[1]
After birth, a diagnosis is often suspected based on the presence of characteristic signs and symptoms, which suggest intestinal obstruction. These may include abdominal distension, vomiting bile, and failure to pass stool. Imaging studies can then be ordered to confirm the diagnosis.[1]
Plain abdominal x-rays are done; they may reveal dilated loops of small bowel with air-fluid levels and a paucity of air in the colon and rectum. A barium enema reveals a microcolon (due to disuse).
Because about 10% of patients also have cystic fibrosis (nearly 100% if meconium ileus is also present), testing for cystic fibrosis should be done.
Intestinal atresias are often discovered before birth: either during a routine sonogram which shows a dilated intestinal segment due to the blockage, or by the development of polyhydramnios (the buildup of too much amniotic fluid in the uterus). These abnormalities are indications that the fetus may have a bowel obstruction which a more detailed ultrasound study can confirm.
Some fetuses with bowel obstruction have abnormal chromosomes. An amniocentesis is recommended because it can determine not only the sex of the baby, but whether or not there is a problem with the chromosomes.
Intestinal obstructions are increasingly being identified through prenatal ultrasounds. This imaging technique may indicate excess amniotic fluid (polyhydramnios), which is caused by the failure of the intestine to properly absorb amniotic fluid. If your physician suspects intestinal atresia or stenosis, your infant will undergo the following diagnostic procedures after being stabilized:
Abdominal X-ray — In most cases, this can establish a diagnosis.
Lower gastrointestinal (GI) series — This is a procedure that examines the rectum, large intestine and lower part of the small intestine. An X-ray contrast agent is given into the rectum as an enema; this coats the inside of the intestines, allowing them to be seen on an X-ray. An abdominal X-ray may show narrowed areas (strictures), obstructions, the width (caliber) of the bowel and other problems.
Upper GI series — This procedure examines the organs of the upper part of the digestive system. It is particularly useful in cases where there is an upper intestinal obstruction (pyloric or duodenal atresia). A liquid called barium, which shows up well on X-rays, is given orally or administered through a small tube placed through the mouth or nose into the stomach. X-rays are then taken to evaluate the digestive organs.
Abdominal ultrasound — Ultrasonography is an imaging technique used to view internal organs as they function, and to assess blood flow through various vessels. Gel is applied to the abdomen and a special wand called a transducer is placed on the skin. The transducer sends sound waves into the body that bounce off organs and return to the ultrasound machine, producing an image on the monitor. A picture or videotape of the test is also made so it can be reviewed later.
Due to the high percentage of infants born with intestinal atresia who also have associated, life-threatening abnormalities,echocardiography and other imaging studies of the cardiac and renal regions may also be performed after the infant is stabilized.
Treatment
Jejunal atresia is typically treated with surgery as soon as possible. Total parenteral nutrition (TPN) is generally necessary for a period of time following surgery until normal meals are tolerated.[3][2]
Fetal and neonatal intestinal atresia are treated using laparotomy after birth. If the area affected is small, the surgeon may be able to remove the damaged portion and join the intestine back together. In instances where the narrowing is longer, or the area is damaged and cannot be used for period of time, a temporary stoma may be placed.
Preoperative management of jejunoileal atresia consists of placing an NGT, giving nothing by mouth, and providing IV fluids.
Surgical repair is the definitive therapy. During surgery, the entire intestine should be inspected for multiple areas of atresia. The atretic portion is resected, usually with a primary anastomosis. If the proximal portion of the ileum is extremely dilated and difficult to anastomose to the distal, unused part of the intestine, it is sometimes safer to do a double-barreled ileostomy and defer anastomosis until the caliber of the distended proximal intestine has diminished.
The prognosis for infants with jejunoileal atresia is very good with > 90% survival. Prognosis is based on the length of remaining small bowel and the presence of the ileocecal valve. Infants who subsequently develop short bowel syndrome require TPN for extended periods. They should be provided continuous enteral feedings to promote gut adaptation, maximize absorption, and minimize the use of TPN. Infants should also be provided small amounts of nutrition by mouth to maintain sucking and swallowing. Prognosis for infants with ultrashort bowel syndrome has improved significantly because of newer surgical techniques including bowel-lengthening procedures (eg, serial transverse enteroplasty procedure or STEP), improved medical care, and the ability to do small bowel transplantation (1, 2, 3).
With jejunoileal atresia, the type of surgery depends on the type of atresia, the amount of intestine present and the degree of intestinal dilation. The most common operation involves removal of the blind intestinal segments, and the remaining ends are closed with sutures. Similarly, a narrowed (stenosed) segment of the intestine can be removed and the bowel sutured together, thus establishing intestinal continuity.
- Surgery is performed under general anesthesia with careful monitoring in a warmed operating room
- The type of defect determines the surgical procedure:
- Type I
- An incision is made into the bowel wall at the level of the web, the web removed and the bowel wall sutured (jejunoplasty)
- Alternately the abnormal bowel is resected (removed) and the cut ends of the bowel anastomosed (sewn together)
- Type II – IV – Atretic bowel segment is removed with anastomosis (cut ends sutured together)
Children with intestinal atresia and stenosis need surgery to correct the problem. The type of operation they require depends on where the obstruction is located. Before the operation, a tube is placed into the stomach (through the mouth) to remove the excess fluid and gas. This prevents vomiting and aspiration, as well as providing babies with some relief from the discomfort caused by a swollen abdomen. Nutrients are provided intravenously, to replace what was lost through vomiting.
Babies with intestinal atresia feed through a tube that goes through their nasal passages directly into the stomach. This is left in place until their bowel function returns, a period of time which may vary from a few days to several weeks. Once the intestines can function normally, nutrition is given orally or through a feeding tube passed into the small intestine. During the period of bowel inactivity, nutrition is provided intravenously.
After the surgery, parents can expect their child’s hospital stay to last from one to several weeks, until the child’s diet is enough to provide a good level of nutrition. However, the hospital stay is often longer for premature infants.
Children will require continued regular follow-up to make sure they are growing and developing normally, and to ensure that their intestine is absorbing enough of the nutrients they need.
If a baby has a sufficient length of intestine, and there are no other associated problems, he or she generally does well after recovery. Complications after surgical therapy are rare, but may occur. During immediate postoperative and early recovery period, intestinal contents may leak at the suture line. This can cause an infection in the abdominal cavity and require additional surgery. Possible long-term complications include short gut syndrome, malabsorption syndromes, and segments of intestine that cannot function properly because they are dilated or paralyzed.
0 200
No Comments