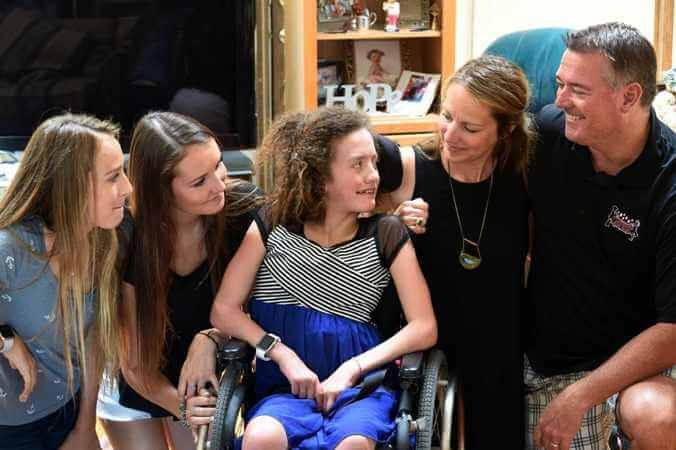
Giant Axonal Neuropathy
Giant axonal neuropathy is a rare, autosomal recessive neurological disorder that causes disorganization of neurofilaments. Neurofilaments form a structural framework that helps to define the shape and size of neurons and are essential for normal nerve function.
Giant axonal neuropathy (GAN) is a rare inherited genetic disorder that affects both the central and peripheral nervous systems. The majority of children with GAN will begin to show symptoms of the disease sometime before five years of age. Signs of GAN usually begin in the peripheral nervous system, which controls movement and sensation in the arms, legs, and other parts of the body.
Giant axonal neuropathy is an inherited condition characterized by abnormally large and dysfunctional axons called giant axons. Axons are specialized extensions of nerve cells (neurons) that transmit nerve impulses. Symptoms of the disorder first become apparent in the peripheral nervous system, in which long axons connect the brain and spinal cord (central nervous system) to muscles and to sensory cells that detect sensations such as touch, pain, heat, and sound. However, axons in the central nervous system are affected as well.
The typical symptoms of GAN are clumsiness and muscle weakness that progresses from a “waddling gait” to a pronounced difficulty in walking. Additional symptoms include numbness or lack of feeling in the arms and legs, seizures, nystagmus (rapid back and forth movement of the eyes), and impaired cognitive development. A characteristic sign of the disease is dull, tightly curled hair that is markedly different from the parents’ in color and texture.
Researchers have discovered more than 20 different mutations associated with GAN in a gene, GAN1, which makes a protein called gigaxonin. These mutations disrupt the regulation or production of gigaxonin in the nervous system. As a result, axons, which are the long tails of neurons that allow them to communicate with other nerve cells, swell up with tangled filaments and become abnormally large. Eventually these axons deteriorate and cause problems with movement and sensation since neurons are no longer able to communicate with each other.
Doctors diagnose GAN by using several tests, including one that measures nerve conduction velocity, a brain MRI, and a peripheral nerve biopsy (in which a bit of tissue from a peripheral nerve is removed and examined to look for swollen axons). A definitive diagnosis using genetic testing is available on a research basis only.
GAN is inherited in an autosomal recessive pattern, which means that both parents of a child with GAN have to carry a copy of the mutated gene. Parents, typically, will show no signs of the disease.
The signs and symptoms of giant axonal neuropathy generally begin in early childhood and get worse over time. Most affected individuals first have problems with walking. Later they may lose sensation, strength, and reflexes in their limbs; experience difficulty coordinating movements (ataxia); and require wheelchair assistance. Visual and hearing problems may also occur. Many individuals with this condition have extremely kinky hair as compared to others in their family.
It is possible that the main title of the report Giant Axonal Neuropathy is not the name you expected. Please check the synonyms listing to find the alternate name(s) and disorder subdivision(s) covered by this report.
Giant axonal neuropathy can also impact the autonomic nervous system, which controls involuntary body processes. Affected individuals may experience problems with the release of urine (neurogenic bladder), constipation, heat intolerance, and reduction in or loss of the ability to sweat.
As the disorder worsens, paralysis, seizures, and a gradual decline in mental function (dementia) can also occur. Most people with giant axonal neuropathy do not survive past their twenties.
General Discussion
Giant axonal neuropathy is a rare neuropathy that severely affects the peripheral as well as the central nervous system. The first symptoms appear in early childhood. This disorder is characterized by abnormalities in the peripheral and central nervous systems including low muscle tone (hypotonia), muscle weakness, decreased reflexes, impaired muscle coordination (ataxia), seizures and intellectual disability. Pale, tightly curled hair is frequently seen in those affected. Giant axonal neuropathy follows autosomal recessive genetic inheritance.
Genetic
Giant axonal neuropathy is caused by mutations in the GAN gene, which provides instructions for making a protein called gigaxonin. Gigaxonin is part of the ubiquitin-proteasome system, which is a process that identifies and gets rid of excess or damaged proteins within cells. In particular, gigaxonin plays a role in the breakdown of neurofilaments, which comprise the structural framework that establishes the size and shape of axons.
The GAN gene mutations that have been identified in people with giant axonal neuropathy result in an unstable gigaxonin protein that breaks down more easily than normal, resulting in much less gigaxonin in cells. In neurons without enough functional gigaxonin, neurofilaments that would otherwise have been broken down by the ubiquitin-proteasome system accumulate. The neurofilaments become densely packed in the giant axons of people with giant axonal neuropathy. These giant axons do not transmit signals properly and eventually deteriorate, resulting in the death of neurons. The loss of nerve cells leads to problems with walking and sensation in people with giant axonal neuropathy.
Symptoms
Symptoms of giant axonal neuropathy occur in early childhood before the age of seven years. Both the central and peripheral nervous systems are affected. The central nervous system includes the brain and spinal cord and the peripheral nervous system spreads out from the brain and spinal cord to all other areas of the body. Characteristics include low muscle tone (hypotonia), muscle weakness, decreased reflexes, impaired muscle coordination (ataxia), seizures and intellectual disability.
In contrast to purely peripheral neuropathies, the reflex of the toes known as Babinski’s sign is often positive, indicating involvement of central motor pathways. Most affected children have pale, tightly curled hair unlike their parent’s hair. Cranial nerves may also be affected leading to facial weakness, abnormal eyes and poor vision. An unusual leg posture is present in some affected children.
Skeletal abnormalities such as scoliosis and foot deformities have been described and are thought to be a result of the nervous system dysfunction. Mental development is in most cases initially normal, but later in childhood degenerative mental changes (dementia) may occur as the disorder progresses. Giant axonal neuropathy is rapidly progressive, usually leading to dependence on a wheel chair by the second decade of life and death in the second or third decade.
Prognosis
GAN generally progresses slowly as neurons degenerate and die. Most children have problems with walking in the early stages of the disorder. Later they may lose sensation, coordination, strength, and reflexes in their arms and legs. As time goes on, the brain and spinal cord may become involved, causing a gradual decline in mental function, loss of control of body movement, and seizures. Most children become wheelchair dependent in the second decade of life.
The prognosis varies but is usually poor. Most patients become wheelchair-dependent in the second decade of life and eventually progress to a bedridden state in early adulthood. Secondary complications, such as respiratory failure, may occur. Life expectancy does not exceed the third decade. Lack of curly hair is correlated with milder disease and slower disease progression, which would suggest a less stark prognosis.
Diagnosis
Diagnosis of giant axonal neuropathy is made by clinical findings and specialized tests including nerve conduction velocity, brain MRI and peripheral nerve biopsy. The hallmark finding on a peripheral nerve biopsy is the appearance of “giant axons” which are caused by the accumulation of neurofilaments. Molecular genetic testing for abnormalities in the GAN gene is available to confirm the diagnosis. Negative mutation screening of the region of the GAN gene which encodes the protein does not exclude the diagnosis of giant axonal neuropathy.
Giant axonal neuropathy usually appears in infancy or early childhood, and is progressive. Early signs of the disorder often present in the peripheral nervous system, causing individuals with this disorder to have problems walking. Later, normal sensation, coordination, strength, and reflexes become affected. Hearing or vision problems may also occur. Abnormally kinky hair is characteristic of giant axonal neuropathy, appearing in almost all cases. As the disorder progresses, central nervous system becomes involved, which may cause a gradual decline in mental function, loss of control of body movement, and seizures.
Causes
Giant axonal neuropathy is an autosomal recessive genetic disorder. This condition is caused by an abnormality in the GAN gene located on chromosome 16 at 16q24.1 that codes for the gigaxonin protein. The abnormal gigaxonin protein causes a portion of the nerve cell called the axon to swell up with deposits of tiny threads of protein called neurofilaments, giving the appearance of giant axons. The giant axons cause degeneration and abnormal functioning of the peripheral nervous system.
Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome16q24.1” refers to band 24 on the long arm of chromosome 16. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits an abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
All healthy individuals carry 4-5 recessive abnormal genes. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Suggestive Findings
The diagnosis of giant axonal neuropathy (GAN) is suggested in individuals with the following:
- Severe early-onset peripheral motor and sensory neuropathy. Nerve conduction studies often show normal to moderately reduced nerve conduction velocity (NCV) but severely reduced compound motor action potentials and absent sensory nerve action potentials.
- Tightly curled lackluster hair that differs markedly from that of the parents. Note: Microscopic examination of unstained hair shows abnormal variation in shaft diameter and twisting (pili torti) similar to the abnormality seen in Menkes disease (see ATP7A-Related Copper Transport Disorders). The hair in individuals with GAN also shows longitudinal grooves on scanning electron microscopy [Kennerson et al 2010, Kaler 2011, Yi et al 2012].
- Central nervous system involvement including intellectual disability, cerebellar signs (ataxia, nystagmus, dysarthria), and pyramidal tract signs
- White matter abnormalities on brain MRI. High signals on T2 sequences in the anterior and posterior periventricular regions as well as the cerebellar white matter are often seen Demir et al 2005.
Establishing the Diagnosis
The diagnosis of GAN is established in a proband by identification of either biallelic pathogenic variants in GAN, the gene encoding the protein gigaxonin or decreased amounts of gigaxonin on immunodiagnostic testing.
Molecular genetic testing
- One genetic testing strategy is sequence analysis of GAN, followed by deletion/duplication analysis if only one or no pathogenic variant is found.
- An alternative genetic testing strategy is use of a multi-gene panel that includes GAN and other genes of interest (see Differential Diagnosis). Note: The genes included and the methods used in multi-gene panels vary by laboratory and over time.
Treatment
Treatment is symptomatic. Children with GAN and their families usually work with a medical team that includes a pediatric neurologist, orthopedic surgeon, physiotherapist, psychologist, and speech and occupational therapists. The major goals of treatment are to maximize intellectual and physical development and minimize their deterioration as time passes. Many children with GAN begin with normal intellectual development and are able to attend a regular school program. Children should be monitored at least once a year to assess their intellectual abilities and to look for the presence of neurological deterioration.
Standard Therapies
Treatment
Treatment of giant axonal neuropathy is symptomatic and supportive and often involves a team of professionals including pediatric neurologists, orthopedic surgeons, physiotherapists, psychologists and speech and language therapists. Services for visually and/or mobility impaired people may be of assistance to people with giant axonal neuropathy. Genetic counseling will be of benefit for patients and their families.
Treatment is symptomatic, focusing on stimulating intellectual and physical development, as well as communication skills, through physical, occupational and speech therapy, since the initiation of symptoms. Skeletal deformities and ophthalmoplegia may need surgery. Intrathecal administration of an AAV9-based gene therapy to restore GAN expression is currently being explored.
Investigational Therapies
Information on current clinical trials is posted on the Internet at www.clinicaltrials.gov. All studies receiving U.S. government funding, and some supported by private industry, are posted on this government web site.
Treatment of Manifestations
Treatment, focused on managing the clinical findings, often involves a team including (pediatric) neurologists, orthopedic surgeons, physiotherapists, psychologists, and speech and occupational therapists. Major goals are to optimize intellectual and physical development and, later in life, to slow the inevitable deterioration of these capacities.
Treatment includes the following:
- Speech and occupational therapy to improve communication and activities of daily living
- Early intervention and special education directed to the individual’s disability. Frequent reassessment is needed because of the progressive nature of the disorder. Special education often becomes necessary between ages five and 12 years.
- Physiotherapy (typically for distal weakness, ataxia, and spasticity) to preserve mobility as long as possible
- Orthopedic surgery as required for foot deformities (Note, however, that most affected individuals become wheelchair bound between ages ten and 20 years for other reasons.)
- Appropriate ophthalmologic treatment (e.g., surgery or glasses), especially if diplopia occurs
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Prognosis
GAN generally progresses slowly as neurons degenerate and die. Most children have problems with walking in the early stages of the disorder. Later they may lose sensation, coordination, strength, and reflexes in their arms and legs. As time goes on, the brain and spinal cord may become involved, causing a gradual decline in mental function, loss of control of body movement, and seizures. Most children become wheelchair dependent in the second decade of life. Some children may survive into early adulthood.
Clinical Characteristics
Clinical Description
Giant axonal neuropathy (GAN) is a neurodegenerative disorder affecting both the peripheral and central nervous systems. GAN is classified within the hereditary motor and sensory neuropathies.
GAN typically begins before age five years and progresses to death, usually by early adulthood. Milder forms of the disease have been reported with later age of onset, extended survival, or modest deterioration of central nervous system [Ben Hamida et al 1990, Zemmouri et al 2000]. Individuals present with a motor and sensory peripheral neuropathy that may also involve the cranial nerves, resulting in facial weakness, optic atrophy, and ophthalmoplegia. Tendon reflexes are often absent; Babinski’s sign may be present as a result of CNS involvement.
The majority of affected individuals show signs of CNS involvement including intellectual disability, cerebellar signs (ataxia, nystagmus, dysarthria), epileptic seizures, and signs of pyramidal tract damage.
Most affected individuals have characteristic tightly curled lackluster hair, unlike their parents.
Most affected individuals become wheelchair dependent in the second decade of life and die in the third decade. They eventually become bedridden with severe polyneuropathy, ataxia, and dementia. Death results from secondary complications, such as respiratory failure.
Other findings. Auditory brain stem evoked responses, visual evoked responses, and somatosensory evoked responses are often abnormal.
EEG often shows increased slow wave activity.
Neuroimaging. Brain MRI and magnetic resonance spectroscopy (MRS) in an individual age 11 years revealed evidence of significant demyelination and glial proliferation in the white matter, but no neuroaxonal loss Alkan et al 2003.
Histopathology
Peripheral nerve biopsy exhibits reduced density of nerve fibers and the presence of giant axons (i.e., distorted nerve fibers with large axonal swellings ≤50 µm) .
Ultrastructural examination of giant axons reveals severe disorganization of neurofilaments (NFs), including loss of parallel orientation along the axons and abnormal clumping .
Reduction of myelin thickness in giant axons, onion-bulb formation by multiple Schwann cell processes, and segmental demyelination and remyelination may also suggest Schwann cell dysfunction.
Note: Giant axons and NF accumulation, initially described as specific hallmarks for GAN, are now known to occur in several forms of the peripheral neuropathy, including the Charcot-Marie-Tooth disease forms CMT2E and CMT4C.
Thus, peripheral nerve biopsy examination is not sufficient to establish the diagnosis of GAN.
Central nervous system. Giant axons are also observed in the cerebral cortex and other parts of the brain in persons with GAN.
Pathophysiology
Structural examination of giant axons often reveals exclusion of mitochondria, endoplasmic reticulum vesicles, and microtubules (MTs) from NF-enriched regions.
and non-neuronal cells. Those alterations extend to GFAP, NFs, keratin, desmin, and vimentin in human and suggest a key role for gigaxonin in maintaining IF architecture.
Skin-derived primary fibroblasts of affected individuals revealed abnormal aggregation of vimentin as an ovoid mass visible on electron or light microscope examination . This human-derived cell type represents a valuable cellular model to study IF organization in GAN. Studies on multiple fibroblasts revealed that vimentin aggregation is partial and conditional, is aggravated upon MT destabilization , and does not depend on TBCB, a partner of gigaxonin.
For more information visit us our website: https://www.healthinfi.com
0 200
No Comments