BH4 Deficiency
Tetrahydrobiopterin deficiency is a rare disorder characterized by a shortage (deficiency) of a molecule called tetrahydrobiopterin or BH4. This condition alters the levels of several substances in the body, including phenylalanine. Phenylalanine is a building block of proteins (an amino acid) that is obtained through the diet. It is found in foods that contain protein and in some artificial sweeteners. High levels of phenylalanine are present from early infancy in people with untreated tetrahydrobiopterin deficiency. This condition also alters the levels of chemicals called neurotransmitters, which transmit signals between nerve cells in the brain.
Infants with tetrahydrobiopterin deficiency appear normal at birth, but medical problems ranging from mild to severe become apparent over time. Signs and symptoms of this condition can include intellectual disability, progressive problems with development, movement disorders, difficulty swallowing, seizures, behavioral problems, and an inability to control body temperature.
Tetrahydrobiopterin deficiency (THBD, BH4D), also called THB or BH4 deficiency, is a rare metabolic disorder that increases the blood levels of phenylalanine. Phenylalanine is an amino acid obtained through the diet. It is found in all proteins and in some artificial sweeteners. If tetrahydrobiopterin deficiency is not treated, excess phenylalanine can build up to harmful levels in the body, causing intellectual disability and other serious health problems.
High levels of phenylalanine are present from infancy in people with untreated tetrahydrobiopterin (THB, BH4) deficiency. The resulting signs and symptoms range from mild to severe. Mild complications may include temporary low muscle tone. Severe complications include intellectual disability, movement disorders, difficulty swallowing, seizures, behavioral problems, progressive problems with development, and an inability to control body temperature.It was first characterized in 1975.
Tetrahydrobiopterin (BH4) deficiencies is a general term for a group of disorders characterized by abnormalities in the creation (biosynthesis) or regeneration of tetrahydrobiopterin, a naturally-occurring compound that acts as a cofactor. A cofactor is a non-protein substance in the body that enhances or is necessary for the proper function of certain enzymes. When tetrahydrobiopterin is deficient, the chemical balance within the body is upset. In most of these disorders, there are abnormally high levels of the amino acid phenylalanine (hyperphenylalaninemia).
Amino acids such as phenylalanine are chemical building blocks of proteins and are essential for proper growth and development. Most of these disorders also cause abnormally low levels of neurotransmitters. Neurotransmitters are chemicals that modify, amplify or transmit nerve impulses from one nerve cell to another, enabling nerve cells to communicate. These chemical imbalances can ultimately cause a wide variety of symptoms and physical findings including progressive neurological abnormalities, lack of muscle tone (hypotonia), the overproduction of saliva (hypersalivation), loss of coordination, abnormal movements, and/or delayed motor development.
The specific symptoms can vary dramatically from one person to another and can range from mild to severe in expression. Prompt diagnosis and treatment of these disorders can prevent potentially severe, irreversible neurological damage. Tetrahydrobiopterin deficiency is caused by mutations in specific genes that encode enzymes required for the biosynthesis or regeneration of tetrahydrobiopterin. Most of these mutations are inherited as autosomal recessive traits.
There are four main forms of tetrahydrobiopterin deficiency sometimes referred to as ‘classical’ tetrahydrobiopterin deficiency. They are: guanosine triphosphate cyclohydrolase I (GTPCH) deficiency; 6-pyruvoyl tetrahydropterin synthase (PTPS) deficiency; pterin-4-alpha-carbinolamine dehydratase (PCD) deficiency; and dihydropteridine reductase (DHPR) deficiency.
The first two disorders are defects in tetrahydrobiopterin creation and the latter two are defects in tetrahydrobiopterin regeneration. Sepiapterin reductase deficiency is a related disorder affecting the third step of tetrahydrobiopterin biosynthesis; it differs from the other disorders in that elevated levels of phenylalanine do not develop. GTPCH deficiency can be broken down in the autosomal dominant form, also known as Segawa syndrome or autosomal dominant dopa-responsive dystonia, or the autosomal recessive form, which is covered in this report. NORD has separate, individual reports on sepiapterin reductase deficiency and Segawa syndrome.
In the past, disorders of tetrahydrobiopterin deficiency were referred to as atypical phenylketonuria or malignant phenylketonuria because physicians believed they were forms of phenylketonuria that did not respond to the standard therapy for that disorder. These terms are now considered obsolete because disorders of tetrahydrobiopterin deficiency are now known to be distinct disorders that are treatable with different therapies.
Symptoms
Although researchers have been able to establish distinct syndromes with characteristic or “core” symptoms, much about these disorders is not fully understood. Several factors including the small number of identified cases, the lack of large clinical studies, and the possibility of other genes influencing the development and progression of these disorders prevent physicians from developing a complete picture of associated symptoms and prognosis.
Disorders of tetrahydrobiopterin deficiency can be classified as transient, mild or severe, which is extremely important in determining specifics of therapy, such as the need for neurotransmitter precursors during treatment (see Standard Therapies below). The specific symptoms and severity associated with tetrahydrobiopterin deficiencies can vary greatly from one person to another, even among individuals with the same subtype or among individuals from the same family.
Overall, the symptoms of GTPCH deficiency, PTPS deficiency, and DHPR deficiency are extremely similar. Generally, PCD deficiency is less severe than the other disorders and affected infants often only exhibit temporary abnormalities of muscle tone and delays in motor development. They are, however, at risk for developing type 2 diabetes (MODY) after the age of 9 years.
Tetrahydrobiopterin deficiencies usually present within the first six months of life and can be detected upon newborn screening because of elevated levels phenylalanine. Infants usually appear normal at birth, although some newborns, particularly in PTPS deficiency, may have a low birth weight. Failure to grow and gain weight (failure to thrive) may occur. Microcephaly, a condition defined as having a head circumference smaller than normally would be expected based on age and gender, is a common finding.
In the severe forms, common, but variable symptoms include neurological dysfunction including convulsions or seizures, swallowing difficulties, poor muscle tone of the trunk of the body (truncal hypotonia), and excess muscle tone of the arms and legs so that they may be stiff and difficult to move (limb hypertonia). Abnormal movements are common and can include abnormal slowness of movement (bradykinesia), rapid, involuntary, purposeless (chorea), slow, involuntary, writhing movements (athetosis), a type of spasm in which the head and feet bend the backward and the back arches (opisthotonus).
Affected children may also exhibit delays in reaching developmental milestones (developmental delays), delays in acquiring skills that require the coordination of mental and physical activities (psychomotor retardation), and, in some cases, intellectual disability.
Neurological dysfunction is progressive and, during the school years, affected individuals may appear uncoordinated or clumsy such as having an abnormal manner of walking (gait abnormalities). In some cases, this clumsiness is due, in part, to involuntary muscle contractions that force the body into abnormal, sometimes painful, movements and positions (dystonia).
Some affected individuals may develop abnormal movements of the eyes that can range from brief upward rolling of the eyes to oculogyric crises, in which the eyes roll upward for a sustained period of time. Sometimes, the eyes may roll downward or move toward each other (converge). Severe oculogyric crises can be associated with additional symptoms including the formation of tears (lacrimation), eye blinking, widening (dilation) of the pupils, drooling, backward flexion of the neck, restlessness, or a general feeling of poor health (malaise).
Additional symptoms that have been reported include excessive production of saliva, lethargy, and irritability. Recurrent episodes of elevated body temperature (hyperthermia) that is not associated with infection may also occur. Certain symptoms may become noticeably worse or more pronounced in the afternoon and evening than in the morning (marked diurnal fluctuation). Swallowing difficulties and poor sucking ability in infants can result poor feeding during infancy.
High levels of phenylalanine are present from early infancy in people with untreated tetrahydrobiopterin deficiency. Infants with this condition appear normal at birth, but medical problems ranging from mild to severe become apparent over time.
The signs and symptoms of this condition can include:
- intellectual disability,
- progressive problems with development,
- movement disorders,
- difficulty swallowing,
- seizures,
- behavioral problems, and
- an inability to control body temperature.
Causes
Tetrahydrobiopterin deficiencies are caused by a mutation in specific genes. Genes provide instructions for creating proteins that play a critical role in many functions of the body. When a mutation of a gene occurs, the protein product may be faulty, inefficient, or absent. Depending upon the functions of the particular protein, this can affect many organ systems of the body, including the brain.
GTPCH deficiency is caused by mutations in the GCH1 gene located on the long arm (q) of chromosome 14 (14q22.1-q22.2) PTPS deficiency is caused by mutations in the PTS gene located on the long arm of chromosome 11 (11q22.3-q23.3). DHPR deficiency is caused by mutations in the QDPR gene located on the short arm (p) of chromosome 4 (4p15.32). PCD deficiency is caused by mutations in the PCBD1 gene located on the long arm of chromosome 10 (10q22.1). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 14q22.1-q22.2” refers to band 22.1-22.2 on the long arm of chromosome 14.
The GCH1 gene provides instructions for creating (encoding) an enzyme called guanosine triphosphate cyclohydrolase I, which is essential in the first of three steps necessary for the creation (biosynthesis) of tetrahydrobiopterin. The PTS gene encodes an enzyme known as 6-pyruvoyl tetrahydropterin synthase, which is essential for the second step in tetrahydrobiopterin biosynthesis. The QDPR gene encodes an enzyme called quinoid dihydropteridine reductase, which is essential for the proper regeneration of tetrahydrobiopterin. The PCBD1 gene encodes for a double-function protein; an enzyme known as pterin-4-alpha-carbinolamine dehydratase, which is also essential for the proper regeneration of tetrahydrobiopterin and for the dimerizing cofactor of hepatocyte nuclear factor 1 (DCoH1).
Mutations in these genes result in low amounts of functional copies of the enzyme that is produced by the specific gene. Consequently, the creation or regeneration of tetrahydrobiopterin is affected, resulting in tetrahydrobiopterin deficiency. Because a PCBD1 gene mutation is rarely associated with severe complications, researchers believe that other enzymes make up for the reduced activity of pterin-4-alpha-carbinolamine dehydratase.
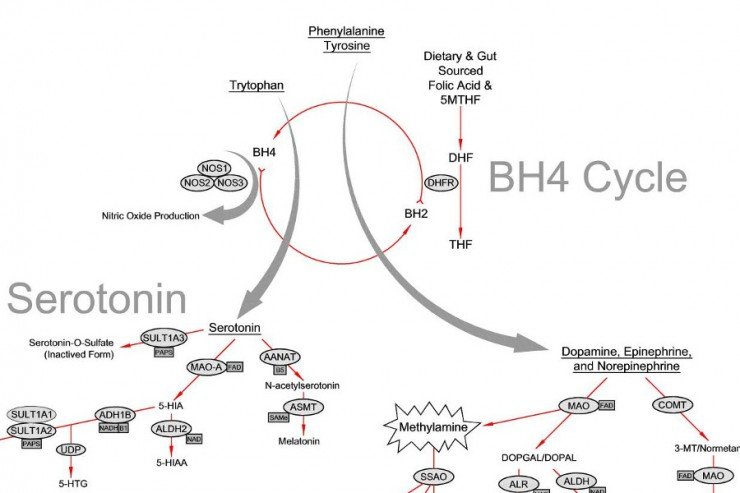
Tetrahydrobiopterin has several functions within the body including breaking down or processing certain amino acids, particular phenylalanine. Phenylalanine is a chemical building block of proteins and is essential for proper growth and development. Tetrahydrobiopterin deficiency results in abnormally elevated levels of phenylalanine (known as hyperphenylalaninemia) in various cells of the body including brain cells. Hyperphenylalaninemia can damage the affected cells, especially brain cells which are particularly sensitive to excess phenylalanine.
Tetrahydrobiopterin is also necessary for the proper development of amine neurotransmitters such as catecholamines (i.e. dopamine, norepinephrine, or epinephrine) and serotonin. Catecholamines are essential for the proper function of certain processes of the brain especially those that control movement. Serotonin helps to regulate mood, appetite, memory, sleep cycles, and certain muscular function. Lack of tetrahydrobiopterin results in a lack of these critical neurotransmitters.
The mutations that cause the forms of tetrahydrobiopterin deficiency discussed in this report are inherited as autosomal recessive traits. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Diagnosis
A diagnosis is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. Disorders of tetrahydrobiopterin deficiency are often found by newborn screening that detects elevated levels of phenylalanine. Further testing is required to distinguish these disorders from other causes of hyperphenylalaninemia such as phenylketonuria, and to determine the specific type of tetrahydrobiopterin deficiency present. Additionally, phenylalanine levels may be normal when a newborn screening is done and can be formal during early infancy, therefore an evaluation for tetrahydrobiopterin deficiencies should be considered in any infant with unexplained neurological symptoms.
Clinical Testing and Workup
Evaluation of urine and dried blood spots (DBS) can measure the levels of pterin metabolites, specifically biopterin or neopterin. Pterins are byproducts of the metabolism of tetrahydrobiopterin. In GTPCH deficiency, levels of biopterin and neopterin are abnormally low. In PTPS deficiency, neopterin is highly elevated and biopterin is very low or absent. In DHPR deficiency, biopterin is highly elevated and neopterin is normal or slightly elevated. In PCD deficiency, neopterin is initially very high, biopterin is slightly reduced, and primapterin is present.
A BH4 loading test, in which infants suspected of tetrahydrobiopterin deficiency are administered BH4, may also be performed. These tests help to distinguish these disorders from the more common phenylketonuria. Elevated phenylalanine levels will drop following a BH4 loading test. In PKU, this drop is minimal to moderate (BH4-responsive PKU). Infants with DHPR deficiency can be missed with urinary pterins or a BH4 loading test. The activity of the enzyme, DHPR, can be measured (enzyme assay) in DBS taken during newborn screening. Reduced activity levels can indicate or confirm a diagnosis of DHPR deficiency. Pterins, neurotransmitter metabolites and folates can be measured in cerebrospinal fluid (CSF). These tests can help to distinguish tetrahydrobiopterin deficiencies from one another and to assess the potential severity of the disease.
Molecular genetic testing can confirm a diagnosis of these disorders. Molecular genetic testing can detect mutations in the specific genes known to cause tetrahydrobiopterin deficiency, but is available only as a diagnostic service at specialized laboratories. The test is often expensive and often not necessary to confirm a diagnosis of a disorder of tetrahydrobiopterin deficiency.
Treatment
Treatment of THB deficiencies consists of THB supplementation (2–20 mg/kg per day) or diet to control blood phenylalanine concentration and replacement therapy with neurotransmitters precursors (L-DOPA and 5-HTP) and supplements of folinic acid in DHPR deficiency.[3]
Tetrahydrobiopterin is available as a tablet for oral administration in the form of tetrahydrobiopterin dihydrochloride (BH4*2HCL)[4]. BH4*2HCL is FDA approved under the trade name Kuvan. The typical cost of treating a patient with Kuvan is $100,000 per year.[5] BioMarin holds the patent for Kuvan until at least 2024, but Par Pharmaceutical has a right to produce a generic version by 2020.[6] BH4*2HCL is indicated at least in tetrahydrobiopterin deficiency caused by GTPCH deficiency or PTPS deficiency.
Prompt recognition and early treatment of tetrahydrobiopterin deficiency is critical in reducing or preventing the potentially severe, irreversible neurologic damage that can occur in severe cases. For GTPCH deficiency, PTPS deficiency, and DHPR deficiency the focus of treatment is to control the level of phenylalanine in the body and restore the proper balance of neurotransmitters in the brain. PCD deficiency may not require any treatment, or may require treatment with synthetic BH4 (tetrahydrobiopterin) temporarily in symptomatic infants or children. Many affected individuals with be treated in a specialty metabolic clinic, where they are seen by physicians with experience in treating these types of disorders.
A diet that limits phenylalanine intake is recommended only in DHPR deficiency, but may not be sufficient on its own in other BH4 disorders. Treatment for individuals with GTPCH deficiency and PTPS deficiency requires oral doses of synthetic tetrahydrobiopterin (BH4; sapropterin dihydrochloride). Individuals with DHPR deficiency may require additional therapy with folinic acid to prevent central nervous system folate deficiency.
Treatment will also require restoring neurotransmitter balance. Affected individuals may be treated with a regimen of amine neurotransmitter precursors, which are substances that are converted into specific neurotransmitters by enzymes in the blood and brain. Specific precursors used to treat tetrahydrobiopterin deficiency are 5-hydroxytryptophan and levodopa (L-dopa) along with carbidopa. In most cases, supplemental therapy with neurotransmitter precursors is required for life.
0 200
No Comments