Bilateral Acoustic Neurofibromatosis
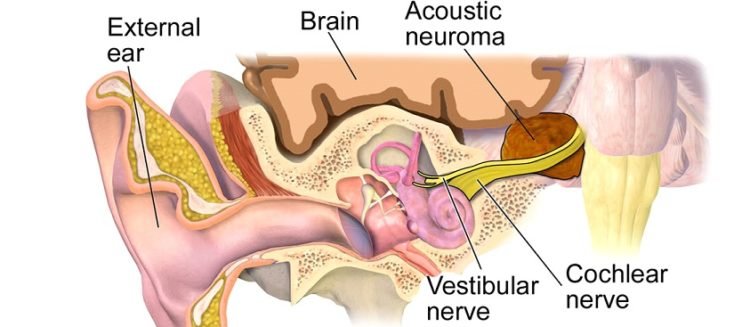
Neurofibromatosis type 2 is a disorder characterized by the growth of noncancerous tumors in the nervous system. The most common tumors associated with neurofibromatosis type 2 are called vestibular schwannomas or acoustic neuromas. These growths develop along the nerve that carries information from the inner ear to the brain (the auditory nerve). Tumors that occur on other nerves are also commonly found with this condition.
The signs and symptoms of neurofibromatosis type 2 usually appear during adolescence or in a person’s early twenties, although they can begin at any age. The most frequent early symptoms of vestibular schwannomas are hearing loss, ringing in the ears (tinnitus), and problems with balance. In most cases, these tumors occur in both ears by age 30.
If tumors develop elsewhere in the nervous system, signs and symptoms vary according to their location. Complications of tumor growth can include changes in vision, numbness or weakness in the arms or legs, and fluid buildup in the brain. Some people with neurofibromatosis type 2 also develop clouding of the lens (cataracts) in one or both eyes, often beginning in childhood.
Neurofibromatosis 2 is a dominantly inherited tumor predisposition syndrome caused by mutations in the NF2 gene on chromosome 22. Affected individuals inevitably develop schwannomas typically affecting both vestibular nerves leading to deafness. Rehabilitation with brainstem implants is improving this outcome. Schwannomas also occur on other cranial nerves, on spinal nerve roots, and on peripheral nerves.
Meningiomas and ependymomas are other tumor features. In excess of 50% of patients represent new mutations and as many as one third are mosaic for the underlying disease causing mutation. Although truncating mutations (nonsense and frameshifts) are the most frequent germline event and cause the most severe disease, single and multiple exon deletions are common.
A strategy for detection of the latter is vital for a sensitive analysis. NF2 represents a difficult management problem with most patients facing substantial morbidity and reduced life expectancy. Surgery remains the focus of current management, although watchful waiting and occasionally radiation treatment have a role. In the future, the development of tailored drug therapies aimed at the genetic level are likely to provide huge improvements for this devastating, life limiting condition.
Neurofibromatosis type II (also known as MISME syndrome – multiple inherited schwannomas, meningiomas, and ependymomas) is a genetic condition which may be inherited or may arise spontaneously. The main manifestation of the condition is the development of symmetric, benign brain tumors in the region of the cranial nerve VIII, which is the “auditory-vestibular nerve” that transmits sensory information from the inner ear to the brain.
Many people with this condition also experience visual problems. NF II is caused by mutations of the “Merlin” gene,[1] which seems to influence the form and movement of cells. The principal treatments consist of neurosurgical removal of the tumors and surgical treatment of the eye lesions.
Historically the underlying disorder has not had any therapy due to the cell function caused by the genetic mutation. However, new drug research and some clinical trials have shown some promise in having beneficial effects. Collaborative research to find better treatments is ongoing, such as the work of the Synodos NF-2 Consortium of scientists.
Neurofibromatosis 2 is a dominantly inherited tumor predisposition syndrome caused by mutations in the NF2 gene on chromosome 22. Affected individuals inevitably develop schwannomas typically affecting both vestibular nerves leading to deafness. Rehabilitation with brainstem implants is improving this outcome.
Schwannomas also occur on other cranial nerves, on spinal nerve roots, and on peripheral nerves. Meningiomas and ependymomas are other tumor features. In excess of 50% of patients represent new mutations and as many as one third are mosaic for the underlying disease causing mutation. Although truncating mutations (nonsense and frameshifts) are the most frequent germline event and cause the most severe disease, single and multiple exon deletions are common.
A strategy for detection of the latter is vital for a sensitive analysis. NF2 represents a difficult management problem with most patients facing substantial morbidity and reduced life expectancy. Surgery remains the focus of current management, although watchful waiting and occasionally radiation treatment have a role. In the future, the development of tailored drug therapies aimed at the genetic level are likely to provide huge improvements for this devastating, life limiting condition.
Central neurofibromatosis, or neurofibromatosis type 2 (NF2), is a genetic disorder marked by the predisposition to develop a variety of tumors of the central and peripheral nervous systems. In contrast to neurofibromatosis type 1 (NF1), NF2 produces a paucity of cutaneous manifestations. (See the image below.)
NORD gratefully acknowledges Kristina Bundra, Pharm. D, NORD Editorial Intern, Madeline Zupan, NORD Editorial Intern from the University of Notre Dame, and D Gareth Evans, MD, FRCP, Director, Neurofibromatosis Clinic, Central Manchester Foundation NHS Trust Regional Genetic Service, St Mary’s Hospital, Manchester, UK, for assistance in the preparation of this report.
Cause
NF II is a microdeletion syndrome involving mutations in the NF2 gene located at 22q12.2 of chromosome 22.[2] It is an inheritable disorder with an autosomal dominant mode of transmission. Incidence of the condition is about 1 in 60,000.[3] There is a broad clinical spectrum known, but all patients checked have been found to have some mutation of the same gene on chromosome 22. Through statistics, it is suspected that one-half of cases are inherited, and one-half are the result of new, de novo mutations.
In some individuals with NF2, the disorder is inherited as an autosomal dominant trait. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females.
In other individuals with NF2, there is no family history of the disease. In such cases, NF2 is caused by new (sporadic) genetic changes (mutations) that appear to occur for unknown reasons.
NF2 results from mutations of a gene located on the long arm of chromosome 22 (22q12.2). Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q”. Chromosomes are further subdivided into bands that are numbered.
The NF2 gene regulates (encodes for) the production of a protein known as merlin/schwannomin that plays a role in suppressing the development of certain tumors (tumor suppressor). According to investigators, merlin/schwannomin is related to a class of proteins (ezrin-radixin-moesin proteins) that serve to link the internal, supportive system within a cell (cytoskeleton) to proteins in cell membranes. Several different mutations of the NF2 gene have been identified in individuals with the disorder (e.g., deletions, nonsense and frameshift mutations). Investigators suggest that different mutations of the gene may contribute to the wide variability of symptoms and findings in affected individuals.
NF2 is a genetic condition. This means that the cancer risk and other features of NF2 can be passed from generation to generation in a family. The gene associated with NF2 is also called NF2. A mutation (alteration) in the NF2 gene, which is a “tumor suppressor,” gives a person an increased risk of developing cancerous and benign tumors and other symptoms of NF2. Most people with NF2 have a mutation in the NF2 gene. Research is ongoing to learn more about the causes of NF2.
Signs & Symptoms
The characteristic symptoms of NF2 usually develop around the time of puberty or during early adulthood. These symptoms may include problems with balance, buzzing or ringing in the ears (tinnitus), and/or gradual hearing loss. These symptoms usually result from the presence of benign tumors on both auditory nerves (acoustic neuromas vestibular schwannomas). Almost all affected individuals develop bilateral vestibular schwannomas by age 30 years
Other tumors of the central nervous system may also develop, and can include neurofibromas, meningiomas, low grade gliomas (mainly benign ependymomas of the spinal cord), and schwannomas. The size, location, and number of tumors may vary in different people affected. (For more information on tinnitus, choose “tinnitus” as your search term in the Rare Disease Database.)
Individuals with NF2 may also develop cloudiness on the lenses of the eyes (posterior capsular cataracts) at a younger age than would otherwise be expected. Symptoms of cataracts may include impaired vision, and, in some cases, the progressive loss of vision. (For more information on this disorder, choose “cataracts” as your search term in the Rare Disease Database.)
People with NF2 generally have fewer brown spots (café-au-lait) on the skin than those who have NF1. Affected individuals may also experience spasms of the facial muscles; generalized muscle weakness, numbness, pain, and/or partial paralysis; difficulty swallowing; and/or impaired speech. Other neurological problems may also develop including headaches and/or seizures.
Diagnosis
The diagnosis of NF2 is confirmed by a thorough clinical evaluation and specialized testing (i.e., CT scan, magnetic resonance imaging (MRI), pneumoencephalogram, or arteriogram are very rarely used nowadays). Molecular genetic testing for mutations in the NF2 gene is available for most affected individuals who have a positive family history.
Treatment
Although the primary method of treating tumors associated with NF2 is surgery, many people with this disease have tumors that grow slowly or not at all. That means they can be closely monitored in an approach called watchful waiting, watch and wait, or active surveillance. In this approach, active treatment would begin if there are signs that the tumor could cause neurological problems or the pattern of growth threatens brain or spinal cord function.
Specifically regarding bilateral acoustic neuromas, a major goal of treatment is to preserve the person’s hearing for as long as possible. However, hearing will eventually be lost at some point. Surgery is usually done in people who can no longer hear or in those with a small tumor where a meaningful attempt can be made at preserving the nerve that makes hearing possible.
In addition, focused radiation therapy is sometimes used for vestibular schwannomas. Like surgery, it carries a risk that the treatment will hurt the person’s hearing in order to control the tumor. The other tumors seen in this disease are usually meningiomas and ependymomas, and these types are typically removed only if they grow to an extent that they put enough pressure on the nearby brain or spinal cord to affect its functioning.
Reports of using bevacizumab (Avastin), a drug that interferes with blood vessel formation to stop tumor growth for treating vestibular schwannomas associated with NF2, have been encouraging. Although only a few people have been treated with this drug, the results include both measureable shrinkage of tumor and partial restoration of hearing loss in some, but not all, patients. This is an experimental treatment and needs more testing, but it holds promise that other drugs may be proven effective as well. Talk with your doctor about clinical trials, which are research studies for new treatments.
0 200
No Comments