
Overview
Agenesis of corpus callosum (ACC) is a rare disorder that is present at birth (congenital). It is characterized by a partial or complete absence (agenesis) of an area of the brain that connects the two cerebral hemispheres. This part of the brain is normally composed of transverse fibers.
The cause of agenesis of corpus callosum is usually not known, but it can be inherited as either an autosomal recessive trait or an X-linked dominant trait. It can also be caused by an infection or injury during the twelfth to the twenty-second week of pregnancy (intrauterine) leading to developmental disturbance of the fetal brain.
Intrauterine exposure to alcohol (Fetal alcohol syndrome) can also result in ACC. In some cases mental retardation may result, but intelligence may be only mildly impaired and subtle psychosocial symptoms may be present.
ACC is frequently diagnosed during the first two years of life. An epileptic seizure can be the first symptom indicating that a child should be tested for a brain dysfunction. The disorder can also be without apparent symptoms in the mildest cases for many years.
Synonyms of Agenesis of Corpus Callosum
- ACC
- Corpus Callosum, Agenesis
Subdivisions of Agenesis of Corpus Callosum
- Acquired Form of ACC
- Aicardi Syndrome
- Autosomal Recessive Inheritance ACC (e.g. Andermann syndrome)
- X-Linked Dominant Inheritance ACC (e.g. ARX)
Spectrum of Abnormalities
ACC may be complete, partial, or atypical. With complete agenesis, the corpus callosum is totally absent.
With partial agenesis (hypoplasia), the anterior portion (posterior genu and anterior body) is formed, but the posterior portion (posterior body and splenium) is not. The rostrum and the anterior/inferior genu are also not formed.
An atypical appearance occurs when the anterior to posterior formation is not respected.
In holoprosencephaly, callosal anomalies are atypical; for example, the splenium may be present without a genu or body. In middle interhemispheric fusion, which is a variety of holoprosencephaly, the genu and splenium may be present without the callosal body.
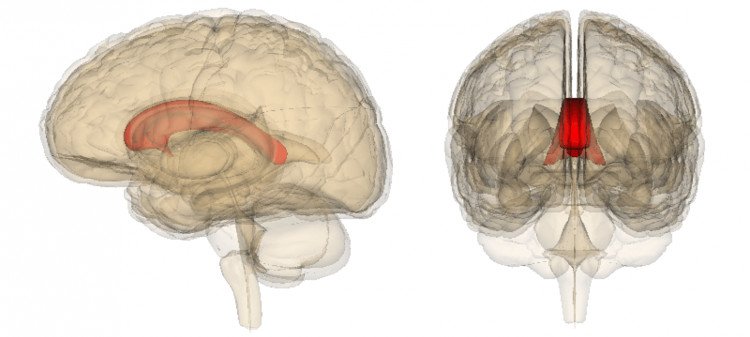
With pseudo–corpus callosum, which involves conditions of complete or partial agenesis, the hippocampal commissure may become enlarged and appear like the posterior part of the corpus callosum.
Secondary destruction of corpus callosum occurs when the genu and anterior body are destroyed, leaving the posterior portion of the corpus callosum intact. This may occur secondary to porencephaly or schizencephaly, as a surgical complication in cases involving the transcallosal approach to the lateral and third ventricle, or with hemisection of the callosum for the treatment of seizures.
Other cerebral malformations may coexist with callosal dysgenesis. Examples of these include interhemispheric cysts; intracranial lipomas; and disorders of neuronal migration, such as neuronal heterotopias, lissencephaly, pachygyria, and, as mentioned, schizencephaly.
Preferred Examination
The diagnosis of callosal agenesis depends on neuroimaging. In the newborn, before closure of the anterior fontanelle occurs, screening ultrasonography (US) may clearly show the absence of the corpus callosum; it may also show parallel lateral ventricles, interhemispheric cysts, hydrocephalus, and other related anomalies. US was the first imaging modality to allow direct sagittal imaging of callosal dysgenesis.
Antenatal diagnosis of ACC is possible from about 20 weeks’ gestation. Characteristic intrauterine US findings include colpocephaly and parallel ventricular walls.
Computed tomography (CT) scan findings are also diagnostic of ACC. Parallel lateral ventricles, colpocephaly, and extension of the third ventricle into the interhemispheric fissure are particularly pertinent findings. In patients with ACC who have an interhemispheric cyst, the preoperative injection of nonionic water-soluble contrast material into the cystic loculations for CT evaluation enables assessment of the ventricular system or of the communication of the cystic components with one another.
Magnetic resonance imaging (MRI) is currently the imaging procedure of choice in infants and children with ACC, even in patients who have previously undergone CT and US examinations.
The multiplanar capability and high soft-tissue contrast that are possible with MRI permit confident diagnosis of ACC and its associated anomalies, especially neuronal migration anomalies or atypical forms of holoprosencephaly. These entities may be extremely subtle or indiscernible on CT or US images.
Agenesis of the corpus callosum may be depicted on CT scanning and US, but MRI is the preferred imaging modality because of its greater sensitivity for depicting associated cerebral anomalies.
Signs & Symptoms
Agenesis of corpus callosum (ACC) may initially become evident through the onset of epileptic seizures during the first weeks of life or within the first two years. However, not all individuals with ACC have seizures. (For more information on these types of seizures choose “epilepsy” as your search term in the Rare Disease Database).
Other symptoms that may begin early in life are feeding problems and delays in holding the head erect. Sitting, standing and walking may also be delayed. Impairment of mental and physical development, and/or an accumulation of fluid in the skull (hydrocephalus) are also symptomatic of the early onset type of this disorder. (For more information, choose “hydrocephalus” as your search term in the Rare Disease Database.)
Non-progressive mental retardation, impaired hand-eye coordination and visual or auditory (hearing) memory impairment can be diagnosed through neurological testing of patients with ACC.
In some mild cases, symptoms may not appear for many years. Older patients are usually diagnosed during tests for symptoms such as seizures, monotonous or repetitive speech, or headaches. In mild cases it may be overlooked due to lack of obvious symptoms during childhood.
Some patients may have deep-set eyes and a prominent forehead. An abnormally small head (microcephaly), or sometimes an unusually large head (macrocephaly), may be present. Tags of skin in front of the ears (pre-auricular skin tags), one or more bent fingers (camptodactyly), and delayed growth have also been associated with some cases of agenesis of corpus callosum.
In other cases wide-set eyes (telecanthus), a small nose with upturned (anteverted) nostrils, abnormally shaped ears, excessive neck skin, short hands, diminished muscle tone (hypotonia), abnormalities of the larynx, heart defects, and symptoms of Pierre-Robin syndrome may be present. (For more information choose “Pierre-Robin” as your search term in the Rare Disease Database).
Aicardi syndrome, thought to be inherited as an X-linked dominant disorder, consists of agenesis of corpus callosum, infantile spasms, and abnormal eye structure.
This disorder is an extremely rare congenital disorder in which frequent seizures, striking abnormalities of the eye’s middle coat (choroid) and retinal layers, and the absence of the structure linking the two cerebral hemispheres (the corpus callosum), accompany severe mental retardation. Only females are affected. (For more information on this disorder, choose “Aicardi” as your search term in the Rare Disease Database).
Andermann syndrome, identified in 1972, is a genetic disorder characterized by a combination of agenesis of corpus callosum, mental retardation, and progressive sensorimotor nervous system disturbances (neuropathy). All known cases of this disorder originate from Charlevois County and the Saguenay-Lac St. Jean area of Quebec, Canada. The gene causing this rare form of ACC was recently identified and testing for this gene (SLC12A6) is currently available.
XLAG (X linked lissencephaly with ambiguous genitalia is a rare genetic disorder in which males have small and smooth brains (lissencephaly), small penis, severe mental retardation and intractable epilepsy. This is caused by mutations in the ARX gene. In females, these same mutations can cause ACC alone, whereas less severe mutations in males can cause mental retardation. Testing for this disorder is also clinically available.
Causes
In most cases, the cause of ACC is unknown. However, agenesis of corpus callosum can be inherited as an autosomal recessive trait or an X-linked dominant trait. This disorder may also be due in part to an infection during pregnancy (intrauterine) leading to abnormal development of the fetal brain.
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy.
The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
In X-linked dominant disorders, a female with only one X chromosome with an abnormal gene will develop the disease. However, the affected male always has a more severe condition. Sometimes, affected males die before birth so that only female patients survive. This seems to be true for one form of agenesis of corpus callosum known as Aicardi syndrome. The majority of patients diagnosed so far have been females. Aicardi syndrome has been seen occasionally in males with an extra X chromosome.
Affected Populations
Agenesis of Corpus Callosum produces symptoms during the first two years of life in approximately ninety percent of those affected. It has been thought to be a very rare condition but the increased use of neuro-imaging techniques, such as MRI, is resulting in an increased rate of diagnosis. This condition may also be identified during pregnancy through an ultrasound. Currently, the highest estimate of incidence is 7 in 1000 individuals.
Related Disorders
Agenesis of corpus callosum can occur in conjunction with spina bifida. Spina bifida is a term meaning open (or non-fused) spine. In spina bifida, one or more of the individual bones of the spine fails to close completely, leaving a cleft or defect in the spinal canal.
Part of the contents of the spine can protrude or herniate through this abnormal opening that produces a meningocele or meningomyelocele. (For more information on this disorder, choose “spina bifida” as your search term in the Rare Disease Database.)
How Is Agenesis of the Corpus Callosum Diagnosed?
Your physician may suspect agenesis of the corpus callosum based on routine prenatal ultrasound exams performed during pregnancy. Your doctor may refer you to our Fetal Medicine Institute for further testing to evaluate your baby’s condition. Tests may include:
High-resolution (level II) ultrasound to confirm the diagnosis
Fetal MRI to help confirm the diagnosis and identify whether associated brain problems are present. This advanced, high-resolution imaging test gives the best information about the underlying cause and the best picture of the rest of the baby’s brain.
MRI after birth to provide further information of your baby
Because agenesis of the corpus callosum may be associated with chromosomal or genetic abnormalities, we may recommend genetic testing by amniocentesis during pregnancy or by blood tests from the baby after birth.
Treatments for Agenesis of the Corpus Callosum
Currently, there are no treatments to restore the corpus callosum to normal. The main course of treatment for agenesis of the corpus callosum is to manage any complications that may arise. Treatment options may include:
- Medications to control seizures
- Speech therapy to help with speech and language development
- Physical therapy to improve muscle strength and coordination
- Occupational therapy to help build self-care and mobility skills such as eating, getting dressed, and walking
- Special education as necessary for cognitive and learning problems
The neurologists and neurosurgeons in the Fetal Brain Program at our Fetal Medicine Institute have extensive experience in the management of agenesis of the corpus callosum. We develop plans that ensure continuity of care from the early stages of pregnancy, after birth, and across childhood.
For more information visit us our website: https://www.healthinfi.com
0 200
1 Comment