Aplasia, Congenital Pure Red Cell
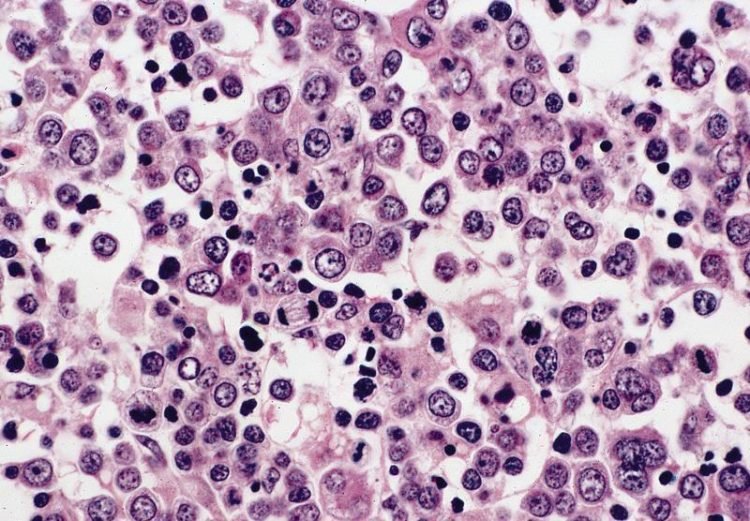
Pure red cell aplasia (PRCA) is an uncommon disorder in which maturation arrest occurs in the formation of erythrocytes. [1, 2] Erythroblasts are virtually absent in bone marrow; however, white blood cell and platelet production are normal. The anemia due to PRCA is usually normocytic but can be macrocytic. In 1922, Kaznelson recognized that this condition was a different entity from aplastic anemia, which presents as pancytopenia.
Acquired Pure Red Cell Aplasia: This is a very rare condition and usually affects adults. It is characterized by an absence of red cell precursors (reticulocytes) in the marrow and a low red blood cell count. The amounts of white blood cells and platelet remain normal.
Transient or Acute Self-limited PRCA: This is the most common type of PRCA. It is identical to acquired PRCA except that, at some point, it simply goes away. Transient PRCA is usually triggered by a virus or drug. In most cases, when the virus clears, or the responsible drugs are eliminated, the PRCA will disappear. This is most dangerous to patients who already have a chronic hemolytic anemia. Patients with otherwise normal functioning bone marrow may even recover without having known they had it.
Inherited or Congenital Pure Red Cell Aplasia (Diamond-Blackfan Anemia): Diamond-Blackfan anemia is a genetic condition usually diagnosed during the first two years of life. About half of patients also have physical malformations or mental retardation. Only several hundred cases have been reported worldwide. The severity of the disease varies by patient.
Relation to bone marrow failure diseases:
The major difference between PRCA and aplastic anemia is that, in PRCA, only the red blood cell line is affected, while the white blood cells and platelets remain at normal levels. In aplastic anemia, all three blood cell types are typically affected.
Acquired pure red blood cell aplasia is a disorder of erythroid precursors that results in an isolated normocytic anemia. White blood cells and platelets are not affected. Symptoms result from anemia and include fatigue, lethargy, decreased exercise tolerance and pallor. Diagnosis requires demonstration of peripheral normocytic anemia and a normocellular bone marrow biopsy with absence of erythroid maturation. Treatment usually involves treatment of underlying cause and in some cases thymectomy or immunosuppression.
Pure red cell aplasia (PRCA) is a rare condition that affects the bone marrow. Bone marrow contains stem cells which develop into the red blood cells that carry oxygen through the body, the white blood cells that fight infections, and the platelets that help with blood clotting. In people with PRCA, the bone marrow makes a reduced number of red blood cells (called anemia). As a result, affected people may experience fatigue, lethargy, and pale skin.[1][2] PRCA has many different causes. A rare congenital form of PRCA, called Diamond Blackfan syndrome, is an inherited condition that is also associated with other physical abnormalities. PRCA can also be due to certain medications, infections, pregnancy, renal failure, and conditions such as thymomas, autoimmune disease (such as systemic lupus erythematosus), cancers of the blood, and solid tumors. In many cases, the cause of the condition is unknown (idiopathic).[1][3] The treatment of PRCA aims to address the underlying cause of the condition and relieve the associated signs and symptoms.
Diagnosis
Pure red cell aplasia in adults can be easily diagnosed when isolated anaemia, in the presence of normal white cell and platelet counts, is associated with a marrow of normal cellularity in which there is an almost complete absence of erythroblasts but normal myeloid cells and megakaryocytes (Dessypris & Lipton, 2004). The classification of the clinical course (acute or chronic) and pathogenesis, such as secondary or idiopathic (no definite underlying disease) is essential to select the optimal therapeutic modality. Evaluations for the possible causes of PRCA should include a previous history of drug use and toxins or infections, liver and kidney functions, immunological examination including auto-antibodies, a bone marrow examination including morphology, chromosome and rearrangement of T cell receptor (TCR) analysis, peripheral-blood flow cytometry, virological examination including parvovirus B19 DNA, and computed tomography and/or magnetic resonance imaging examinations to rule out the presence of thymoma and neoplasms.
Today, a careful assessment of the increase of large granular lymphocytes (LGLs) is especially critical and an analysis of immunophenotype and TCR rearrangement of lymphocytes may be essential for ruling out LGL leukaemia, also referred to as granular lymphocyte proliferative disorders (GLPD) (Oshimi et al, 1993) or lymphoproliferative disease of granular lymphocytes (LDGL) (Go et al, 2001). LGL leukaemia was the most common underlying disease of secondary PRCA in a single institutional study from the United States, and the second most common cause in Japan (Lacy et al, 1996; Mamiya et al, 1997; Sawada et al, 2007). Since the diagnosis of LGL leukemia is somewhat difficult in patients without lymphocytosis, this group of patients can be misdiagnosed as idiopathic PRCA although LGL leukemia-associated PRCA may require a different treatment for the primary disease. It is a heterogeneous disorder characterized by a persistent increase in the number of peripheral blood LGLs, and the majority of patients have a clonal rearrangement of T-cell receptors (Oshimi et al, 1993; Semenzato et al, 1997; Chan et al, 2001). Clonal disorders of LGLs arise from either mature T lymphocytes or NK cells, and may be indolent or behave as an aggressive disease. T-cell LGL leukaemia is the most common form of clonal LGL disorders and most cases behave in an indolent fashion. Neutropenia is the most frequent cytopenia in T-cell LGL leukaemia, and anaemia occurs in 48% of the patients (Loughran, 1993; Lamy & Loughran, 1998, 2003).
The evidence of a granular lymphocytosis greater than 2 × 109/l lasting for more than 6 months has been regarded as the criteria for defining the disease (Loughran & Starkebaum, 1987; Semenzato et al, 1987; Oshimi, 1988). However, the normal range for peripheral blood LGL counts is 0·223 ± 0·099 × 109/l (Loughran et al, 1987) and clonal disease has been documented in 8% of patients when absolute LGL counts are between 0·6 to 1·0 × 109/l (Loughran, 1993). Thus, an expansion of a restricted LGL subset demonstrates the diagnosis of LGL-leukaemia and a 6-month follow-up criterion is not necessary when clonality is established (Semenzato et al, 1997). The characteristic finding is the presence of increased numbers of LGL, usually identified by a greater size than normal lymphocytes, abundant pale cytoplasm, and prominent azurophilic granules. However, these features may vary, even among cells from the same patient (Loughran, 1993). The granulation can range from fine to coarse, and some cells may have otherwise characteristic features but lack granules (sometimes called large agranular lymphocytes) (Bassan et al, 1986). Occasionally, clonally expanded lymphocytes with a characteristic CD3+, CD57+ phenotype may not have LGL morphology on a peripheral smear (Ahern et al, 1990) but may represent in vivo antigen-activated cytotoxic effector T cells. An increase of CD3+/CD56− or CD3−/CD56+ cells by peripheral-blood flow cytometry and/or an inverted CD4+/CD8+ cell ratio (<1·0) suggests the existence of LGL leukaemia (Gonzales-Chambers et al, 1992).
Tests and tools doctors use to diagnose PRCA include:
- Physical exam and history
- Blood tests:
- A complete blood count with a differential that looks at the total number and type of blood cells in your blood
- A reticulocyte count to determine blood levels of young red cells
- A blood smear so your doctor can look at your blood under a microscope
- Bone marrow examination, including aspiration and biopsy to see how your bone marrow looks (required for diagnosis)
Patients should consult a hematologist who has experience with bone marrow failure diseases. If a rheumatoid disorder is responsible, a rheumatologist should also be consulted.
- CBC, reticulocyte count
- Bone marrow examination
Pure RBC aplasia presents with a normocytic anemia but normal WBC and platelet counts. Reticulocytes are decreased. The bone marrow reveals normal cellularity with a maturation arrest at the proerythroblast stage. In parvovirus B19 infection, giant pronormoblasts may be present.
A physician may employ one or more of the following methods to arrive at an accurate diagnosis of Pure Red Blood Cell Aplasia:
- Patient history and physical examination to assess and understand the cause
- Evaluating the function of the extremities and testing how alert the individual is
- Examination of the heart and lungs to identify the presence of any abnormalities
- Tests to check for abnormalities with the immune response
- Tests to evaluate bone marrow function
- Cultures to identify the presence of any infection
Many clinical conditions may have similar signs and symptoms. Your healthcare provider may perform additional tests to rule out other clinical conditions to arrive at a definitive diagnosis.
Signs & Symptoms
Acquired Pure Red Cell Aplasia is characterized by a decrease in the number of red blood cells produced in the bone marrow. Individuals with this disorder are deficient in the number of precursors of red blood cells (erythroblasts). Levels of the hormone erythropoietin that stimulates the bone marrow to produce red blood cells are usually elevated.
Affected individuals may experience fatigue, lethargy, and/or abnormal paleness of the skin (pallor).
- By definition, all PRCA patients have anemia. Symptoms of anemia can include:
- Fatigue
- Pale skin
- Dizziness
- Shortness of breath
Acquired PRCA can be caused by an underlying condition that may add other symptoms.
Symptoms can include:
- pallor
- tachycardia
- decreased activity
Symptoms of pure RBC aplasia are generally mild and relate to the degree of the anemia or to the underlying disorder. The onset of pure red blood cell anemia usually is insidious, often occurring over weeks or months. Symptoms related to anemia include fatigue, lethargy, decreased exercise tolerance and pallor.
The signs and symptoms associated with Pure Red Cell Aplasia may include:
- Generalized weakness in the body
- Changes in skin tone
- Excessive bleeding that may lead to easy bruising
- Heart conditions that may lead to the development of a stroke
- Anemia
- Developing an infection
- Cough
- Changes in eye color (yellow)
- Negative effect on the growth spurt
Causes
Acquired Pure Red Cell Aplasia is thought to be an autoimmune disorder possibly caused either by a tumor of the thymus gland, certain drugs or a viral infection. It is one of a group of bone marrow failure syndromes.
- Autoimmune disease.
- [2]
- Viral infections such as HIV, herpes, parvovirus B19 (Fifth disease),[3] or hepatitis.[citation needed]
- Association of pure red cell aplasia with T-cell large granular lymphocyte leukemia is well recognized, especially in China.[4]
- Many cases of PRCA are considered idiopathic in that there is no discernible cause detected.[5]
- Drugs such as mycophenolic acid[6] or erythropoietin.[7][citation needed]
- The term “hereditary pure red cell aplasia” has been used to refer to Diamond-Blackfan anemia.[8]
Generally, Pure Red Blood Cell Aplasia occurs as a result of an immunological defect/injury to the bone marrow, as a consequence of defective early-phase maturation of erythrocytes (red blood cells).
- It may arise due to a defect at birth too
- PRCA could be acquired over time as a result of an underlying autoimmune condition, such as systemic lupus, and some types of cancer (Example – thymoma)
- Use of certain medications, such as anti-erythropoietin antibodies, anti-seizure medicines, chloramphenicol, etc., could lead to PRCA
- Drug use/abuse is also a reported cause of PRCA
- Viral infections (such as mumps, viral hepatitis, etc.) could lead to a transient form of PRCA as well
In some cases, the cause of PRCA is unknown (idiopathic), with the individual having no prior history or symptoms for it.
Treatment
The treatment goal is to restore red blood cell production and to treat any underlying disorder. The most common treatments are:
- Blood Transfusions to increase red blood cell levels. Patients who don’t respond to other treatments may need to rely on transfusions on an ongoing basis.
- Corticosteroids, like prednisone, are often the initial treatment for PRCA. Unfortunately, the large doses that are often required can have severe side effects. For that reason, corticosteroid medications are rarely used alone as a treatment.
- Immunosuppressive therapy may be used if the underlying cause of the PRCA is immunological and the patient hasn’t responded to corticosteroids. Drug therapies include cyclophosphamide, cyclosporine, azathioprine with or without corticosteroids, and anti-thymocyte globulin. In some cases, rituximab may be effective. PRCA caused by viruses (especially B19 parvovirus) is treated with immunoglobulin infusions. Thymoma-associated PCRA has to be treated with surgical removal of the thymoma. There is a significant relapse rate after immunosuppressive therapy, but treatments can be repeated.
PRCA is considered an autoimmune disease as it will respond to immunosuppressant treatment such as ciclosporin in many patients,[9] though this approach is not without risk.[10]
It has also been shown to respond to treatments with Rituximab and Tacrolimus.
- Immunosuppression
- Sometimes intravenous immunoglobulin (IV Ig) or thymectomy
Pure RBC aplasia has been successfully managed with immunosuppressants (prednisone, cyclosporine, or cyclophosphamide), especially when an autoimmune mechanism is suspected. Pure RBC aplasia secondary to parvovirus infection is treated with intravenous immunoglobulin. Because patients with thymoma-associated pure RBC aplasia improve after thymectomy but are not always cured, CT is used to seek the presence of such a lesion, and surgery is considered.
The main goals of treatment for pure red cell aplasia (PRCA) are to restore the production of red blood cells, maintain adequate hemoglobin levels, and treat underlying disorders that may be causing the condition. The initial treatment plan typically includes blood transfusions for individuals who are severely anemic and have cardiorespiratory failure.[2] PRCA due to medication or infections is usually reversible within a few months. Therefore, medications that may be causing the condition should be discontinued, and infections that may cause the condition should be treated.[2] Underlying conditions that may cause PRCA such as a thymoma, hematological cancers, solid tumors, and systemic lupus erythematosus (SLE) should be treated as necessary as well.[2] When the condition is idiopathic (of unknown cause) or due to an autoimmune disorder, PRCA is typically initially treated with corticosteroids.[2]
It has been reported that individuals who seem to be resistant to treatment may respond to a single course of intravenous immunoglobulin (IVIG,) while others have responded to a single dose. In the United States, financial issues may make it difficult to obtain this treatment because IVIG is expensive and is not approved by the Food and Drug Administration to treat PRCA.[3]
The treatment of PRCA associated with thymoma, B19 parvovirus infection, ABO-incompatible stem cell transplantation, antibody-mediated inhibition induced by rhEpo, or pregnancy will be discussed later. DBA is typically treated with glucocorticoids, usually prednisone. Hematopoietic stem cell transplantation has also been used in patients unresponsive to glucocorticoids at sustainable doses.25 Myelodysplastic primary PRCA should be treated as appropriate for a myelodysplastic syndrome.5 If the patient is using a medication associated with PRCA, and no other PRCA-associated syndrome is present, a trial of drug discontinuation should be considered. If an infectious disorder associated with PRCA is present, specific treatment of that disorder should be initiated. Similarly, if chronic lymphocytic leukemia, Hodgkin or non-Hodgkin lymphoma, or other lymphoproliferative disorders are complicating PRCA, treatment of those disorders should be initiated.
Otherwise, for primary acquired autoimmune PRCA, PRCA with LGL leukemia, PRCA associated with solid tumors, or secondary PRCA refractory to other therapy, the treatment of choice is immunosuppression. In principle, PRCA secondary to autoimmune/collagen vascular disorders may respond to therapy specific to the management of those disorders. In practice, the treatment of these disorders is usually immunosuppression, and in cases where alternative disease-modulating therapy is available, the patient is usually referred to the
hematologist because it was ineffective. The goal of treatment is to attain a normal hemoglobin concentration without any requirement for transfusion; a partial response is attainment of transfusion independence with a low but clinically acceptable hemoglobin concentration.
For more information visit us our website: https://www.healthinfi.com
0 200
No Comments