Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is an acquired, inflammatory eye condition affecting the retina, retinal pigment epithelium (pigmented layer of the retina), and choroid. It usually affects both eyes and is characterized by multiple, yellow-white lesions in the back of the eye.
The condition can significantly impair visual acuity if the macula is involved. APMPPE typically resolves on its own in weeks to months. While the cause is unknown, about a third of cases appear to develop after a flu-like illness. Non-ocular symptoms are uncommon, but cerebral vasculitis can be present and may cause permanent and/or severe neurological complications.
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) is an acquired inflammatory uveitis that belongs to the heterogenous group of white dot syndromes in which light-coloured (yellowish-white) lesions begin to form in the macular area of the retina. Early in the course of the disease, the lesions cause acute and marked vision loss (if it interferes with the optic nerve) that ranges from mild to severe but is usually transient in nature.
APMPPE is classified as an inflammatory disorder that is usually bilateral and acute in onset but self-limiting. The lesions leave behind some pigmentation, but visual acuity eventually improves even without any treatment (providing scarring doesn’t interfere with the optic nerve).
It occurs more commonly in females and is more likely to affect persons between 20 and 30 years of age, but has been seen in people aged 16 to 40. It is known to occur after or concurrently with a systemic infection (but not always), showing that it is related generally to an altered immune system. Recurrent episodes can happen, but are extremely rare.
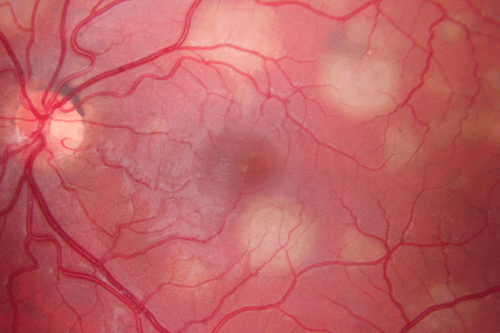
APMPPE is one of the white dot syndromes that occurs in young healthy adults and similarly affects males and females. It is usually bilateral, although may be worse in one eye. APMPPE is generally a self-limited condition that requires no treatment and has a good prognosis.
A disease of the retinal macula seen in otherwise healthy young patients, who develop bilateral acute onset visual disturbance—e.g., scotomas, blurred vision, red eye or floaters, scintillating scotomas, and hemianopic blur; initial acuity varies widely by site of retinal involvement with one-third of eyes having less than 20/100 when first seen.
Aetiology Idiopathic; influenza-like illness.
Systemic defects in APMPPE Erythema nodosum, TB, sarcoidosis, nephritis, thyroiditis, potentially fatal cerebral vasculitis, CSF defects.
Fundoscopy Multiple, yellow-white, flat (placoid) lesions in macula at the level of the retinal pigment epithelium (RPE); lesions are well-circumscribed and discrete; the overlying retina usually appears normal and subretinal fluid is rare; vitritis and iritis are uncommon and mild.
Fluorescein angiography Acute lesions are hypofluorescent, then stain with increasing hyperfluorescence; inactive lesions may fluoresce as window defects due to depigmentation of the RPE.
Prognosis Good; the norm is a rapid regression of the fundus lesions and a delayed return of visual acuity to near normal levels.
Symptoms
The onset of ocular symptoms are usually preceded by episode of viral or flu-like symptoms such as fever, cough or sore throat (however this is not always the case). Patients can typically present erythema nodosum, livido reticularus, bilateral uveitis, and sudden onset of marked visual loss associated with the appearance of multiple lesions in the retina.
These lesions may be colored from grey-white to cream-shaded yellow. Other symptoms include scotomata and photopsia. In weeks to a month times the lesions begin to clear and disappear (with prednisone) leaving behind areas of retinal pigment epithelial atrophy and diffuse fine pigmentation (scarring). Rarely choroidal neovascularization occur as a late onset complication.
APMPPE is characterized by multiple yellow-white placoid (flattened) subretinal lesions of the posterior pole. These lesions block out background choroidal fluorescence during the early stages of Fluorescein Angiography (FA). The late stage of FA demonstrates an accumulation of dye in the diseased RPE.
FA, a diagnostic test in which a dye is injected into the arm and the flow of this dye as it enters the eye is studied, shows a choroidal flush which is normally bright as the rich vasculature of the choroid lights up in normal individuals.
This disease involves the RPE, particularly in the Macula area, causing visual loss with subsequent spontaneous resolution. The lesions described above frequently occur in both eyes and in various stages of evolution, typically resolving in weeks to months and leaving residual pigmentation.
If the Macula is affected, vision may be significantly decreased but recovery is the norm, with 80% of those eyes affected achieving a visual acuity of 20/40 or better.
Photopsia which is the sensation of seeing lights, sparks, or colors and which is caused by diseases of the Retina or brain
Metamorphopsia/micropsia refers to the distorting of vision (such as straight lines appearing wavy) or objects appearing smaller
Photophobia
In rare cases, redness of the Conjunctiva or Episcleritis.
Late stage symptom is commonly mild visual impairment (20/25 to 20/40). Significant visual loss (20/200) is rare.
Prior to onset of the condition, about a third of people have flu-like or viral symptoms such as fever, swollen lymph glands, nausea, vomiting, joint pain and/or tenderness. Headaches may also be present. Rarely, there may be neurological signs such as temporary loss of speech (aphasia) and/or weakness of the arms and legs.
In the early stages of APMPPE, affected people may notice areas of visual blotchiness; flashes of light (photopsia) caused by irritation of the retina; distortion of shapes (metamorphopsia); increased sensitivity to light (photophobia); and/or conjunctivitis. Later, affected people usually develop impairment of vision. In rare cases, vision impairment may be severe.
In most cases, the disorder resolves within a few weeks without permanent loss of visual acuity. However, in some cases, visual acuity does not improve. Neurological symptoms develop in some affected people and should be evaluated and treated promptly, as neurological involvement can result in severe complications.
There are rare cases of chronic or recurrent APMPPE; it has been suggested that these cases represent relentless placoid chorioretinitis. This is a rare condition characterized by the development of multiple inflammatory lesions resembling those seen in APMPPE. However, unlike in APMPPE, the lesions continue to expand in size and number, with a relentless course over many months.
Cause
The cause of the inflammation remains unknown, with various theories of it occurring as an autoimmune response to a mild infection, or the possibility of it being viral because of the preceding flu-like illness that generally accompanies it. It is usually associated with HLA-B7 and HLA-DR2.
No one has discovered the cause of APMPPE, although some clues exist. Approximately 25-30% of patients with APMPPE report a flu-like illness shortly before developing the visual problems, leading many to suspect that an unidentified virus is the cause.
Blood for antibody testing and many cultures have yet to reveal a convincing candidate virus or bacterium. APMPPE is not just an eye disease, as some patients show inflammation in the spinal fluid, inflammation of blood vessels in the brain, rashes, and joint inflammation. Patients presenting with the eye problems of APMPPE should be specifically questioned to see if these other associated problems are present.
Treatment
Owing to the self-limiting nature of the disease, treatment is generally not required. In cases where lesions appear to be interfering with the optic nerve, methyl prednisone is prescribed
The treatment of APMPPE is somewhat controversial, but the general consensus is that no treatment seems to alter the course of the ocular lesions. In cases complicated by subretinal neovascularization (growth of new blood vessels), laser photocoagulation may be useful.
In most cases, the lesions resolve spontaneously and no therapy is required. Some clinicians have used corticosteroids (which suppress inflammation) to treat the ocular findings and any severe systemic involvement. However, there is no evidence that treatment with corticosteroids affects the visual outcome.The use of steroids has also been suggested when treating cases where the macula is involved. Cycloplegics may be useful for severe iritis, which is an uncommon finding.
It is recommended that people with questions about treatment options for themselves or family members speak with their health care provider.
This disease commonly shows rapid healing of the lesions of the Retina over one to two weeks. Symptoms such as vision compromise or visual field defects may persist for up to six months. The vast majority of patients recover to an acuity level between 20/20 to 20/30. Thus, no treatment is used for the retinal findings alone.
If your disease is accompanied by Anterior Uveitis or other eye inflammatory condition, your eye doctor will treat the inflammation appropriately. Your eye doctor should also monitor you for any new blood vessel growth in your choroid as a result of any chronic blood flow deficiency of the choroidal vessels.
If you develop APMPPE and have any signs or symptoms suggestive of a neurological or systemic disorder, make sure your doctor evaluates you carefully for CNS conditions or refers you to a specialist for evaluation for any CNS processes known to be associated with the disease.
Even though APMPPE is generally benign, it can in rare cases be an indicator of serious, possibly life-threatening CNS pathology such as those mentioned above. If your doctor fails to investigate any neurological or body disorder presenting with APMPPE, it can have devastating consequences.
Currently, standard treatment protocols are not well-established for Acute Posterior Multifocal Placoid Pigment Epitheliopathy. However, many cases are known to resolve spontaneously and the condition is self-limiting.
The treatment may include the following measures:
- Oral medications (including pain medications)
- Steroids may be administered to control the inflammation
- Dark glasses may be prescribed for light-sensitivity
- Addressing underlying conditions that may be contributing to progression of the disorder
- If the underlying cause is unknown, then decreasing inflammation is the main step towards treating APMPPE
- Administration of immunomodulators, which are medications to control dysfunctional immune system
It is important to note that steroids may not be used in all cases, since it can worsen the condition. A healthcare provider will provide the best treatment options based upon each individual’s specific circumstances.
Risk Factors
Approximately 33% of patients report a preceding viral or flu-like illness prior to APMPPE symptom onset. APMPPE has been described in cases of thyroiditis, erythema nodosum, granulomatosis with polyangiitis, polyarteritis nodosa, nephritis, sarcoidosis, scleritis, ulcerative colitis, CNS vasculitis, and post-vaccination.
Other infectious associations include group A streptococcus, adenovirus, influenza, hepatitis B, Lyme disease, mumps, and tuberculosis. Genetics may play a role in an individual’s risk for APMPPE as several associations have been reported including HLA-B7 and HLA-DR2 genetic haplotypes. Recurrence is rare, but typically has been reported with worse prognosis.
Diagnostic procedures
No specific laboratory tests exist to confirm the diagnosis. Fluorescein angiography and Indocyanine Green angiography are typically performed. Optical coherence tomography and fundus autoflourescense may also have a role in diagnoses. Characteristic findings of these diagnostic procedures include:
Fluorescein Angiogram (FA):
Early hypofluorescence (blockage) corresponding to the placoid lesions followed by late, irregular hyperfluorecent staining.
Indocyanine Green (ICG) Angiogram:
Early and late hypofluorescence corresponding to the placoid lesions.
Fundus autoflourescense (FAF):
Early and late hypoautoflourescence corresponding to the placoid lesions. Hypoautoflourescence may persist at borders after lesion resolution.
Optical coherence tomography (OCT):
Hyperreflectivity from the outer plexiform layer to the RPE with normal retinal thickness in acute lesions. Hyperreflectivity of outer layers resolve along with resolution of the lesion.
Differential Diagnosis
Other White-Dot syndromes can resemble APMPPE.
These include, but are not limited to:
- Multiple evanescent white dot syndrome
- Serpiginous choroiditis (especially in chronic, recurrent cases)
- Relentless placoid choroiditis
- Multifocal choroiditis and panuveitis
- Punctate inner choroidopathy
- Birdshot chorioretinopathy
Other infectious uveitis (tuberculosis, fungal disease, syphilis), choroidal metastases, and lymphoma may present with placoid lesions as well and should be ruled out with appropriate tests if clinical suspicion high.
- Serpiginous choroiditis
- Multiple evanescent white dot syndrome (MEWDS)
- Birdshot retinochoroidopathy
- Multifocal choroiditis
- Punctate inner choroidopathy
- Acute zonal occult outer retinopathy
- Laser pointer injury
- Uveitis (sarcoid, syphilis, etc)
For more information visit us our website: https://www.healthinfi.com
0 200
No Comments